
Bij uzelf of bij uw kind is de ziekte 'metachromatische leukodystrofie (MLD)' vastgesteld. In deze folder kunt u lezen wat de ziekte inhoudt en welke behandelingen voor deze ziekte worden gegeven. We bespreken ook een aantal problemen waarmee u te maken kunt krijgen. Als u vragen heeft of uw eventuele zorgen wilt bespreken, kunt u altijd contact opnemen met één van de kinderneurologen van het Amsterdam Leukodystrofie Centrum in het Amsterdam UMC, locatie VUmc.
Inhoudsopgave
2. De werking van het zenuwstelsel 3
3. Wat is metachromatische leukodystrofie / MLD? 4
5. Welke klachten of verschijnselen kunnen er door MLD ontstaan? 6
6. Hoe wordt MLD vastgesteld? 8
7. Welke onderzoeken zijn er nog meer nodig? 11
8. Welke behandelingen zijn er voor MLD? 12
Metachromatische leukodystrofie (MLD) is een stofwisselingsziekte. Stofwisseling is een ander woord voor het omzetten en verwerken van stoffen in het lichaam. Dit is te vergelijken met een proces (de stofwisseling) in een fabriek (het lichaam). Hierbij worden bouwstoffen omgezet tot een nieuw product en komt afval vrij dat opgeruimd moet worden. Er zijn verschillende hulpmiddelen (enzymen) nodig om dit proces goed te laten verlopen. Wanneer er een hulpmiddel tekort is of niet goed werkt, kan het proces verstoord raken. Hierdoor kan bijvoorbeeld een nieuw product niet goed worden aangemaakt of afval niet goed worden opgeruimd. Te weinig productaanmaak en opstapeling van afval kunnen schade in de fabriek veroorzaken. Dit is wat er gebeurt bij een stofwisselingsziekte. Doordat er in het lichaam veel stofwisselingsprocessen plaatsvinden, bestaan er ook veel verschillende stofwisselingsziekten. In totaal zijn er ongeveer 600 stofwisselingsziekten bekend.
Bij MLD is er bijna altijd een verlaagde activiteit van het enzym ‘Arylsulfatase A (ASA)’. Dit wordt ook wel een enzymdeficiëntie of ASA-deficiëntie genoemd. Heel soms is er een enzymdeficiëntie van ‘Saposin B’. ASA en Saposin B zijn nodig voor de afvalverwerking van een vettige stof, ook wel ‘sulfatide’ genaamd. Doordat de activiteit van ASA (of Saposin B) verlaagd is, wordt sulfatide onvoldoende opgeruimd. Het overschot aan sulfatide stapelt zich daardoor op in het lichaam. Dit gebeurt in lysosomen, één van de locaties in het lichaam waar afval wordt opgeruimd. MLD wordt dan ook wel een lysosomale stapelingsziekte genoemd. Sulfatide is voornamelijk aanwezig in het zenuwstelsel. Hierdoor vindt de meeste sulfatidestapeling in het zenuwstelsel plaats en ontstaat daar de grootste schade. Schade van het zenuwstelsel kan verschillende klachten veroorzaken. Om dit goed te begrijpen is het belangrijk om te weten hoe het zenuwstelsel werkt.
Het zenuwstelsel regelt alles wat er in ons lichaam gebeurt en wat ons lichaam moet doen. Zo zorgt het zenuwstelsel bijvoorbeeld ervoor dat we kunnen lopen en lezen, dat we kunnen voelen en zien, en dat we weten wanneer we naar het toilet moeten. Dit doet het zenuwstelsel door informatie van binnen en buiten het lichaam om te zetten in elektrische impulsen. Deze elektrische impulsen worden door het hele lichaam vervoerd en op de juiste plaatsen weer omgezet in informatie. Om dit goed te kunnen doen, bestaat het zenuwstelsel uit verschillende onderdelen: de hersenen, het ruggenmerg en de zenuwen. De hersenen en het ruggenmerg worden samen het centrale zenuwstelsel genoemd. De zenuwen worden het perifere zenuwstelsel genoemd.
De hersenen (centrale zenuwstelsel)
De hersenen zijn vergelijkbaar met de directiekamer van de fabriek. In de hersenen komt alle informatie (in de vorm van elektrische impulsen) vanuit het lichaam samen en op basis van deze informatie worden beslissingen genomen. Deze beslissingen bepalen wat de rest van het lichaam uitvoert. Om dit goed te kunnen doen, moet informatie worden begrepen, worden opgeslagen en opnieuw worden geraadpleegd. Dit vindt allemaal plaats in de hersenen en gebeurt razendsnel. Bijvoorbeeld, wanneer informatie over rook vanuit je ogen je hersenen binnenkomt, begrijpen je hersenen dat er brand is (deze informatie is eerder in je hersenen al opgeslagen), en wordt besloten dat je 112 moet bellen. Deze beslissing wordt vanuit je hersenen doorgegeven aan de rest van je lichaam. Dit gebeurt via het ruggenmerg en de zenuwen.
Het ruggenmerg (centrale zenuwstelsel)
Het ruggenmerg bestaat uit meerdere grote zenuwbanen. De functie van die zenuwbanen kan worden vergeleken met de functie van procesleiders. De procesleiders staan in direct contact met de directiekamer (de hersenen). Wanneer in de directiekamer een beslissing is genomen, geven de procesleiders deze informatie (in de vorm van elektrische impulsen) door aan een zenuw. Deze zenuw is verbonden met een specifieke plaats in het lichaam. Daarnaast ontvangen de procesleiders van deze zenuwen ook informatie uit plaatsen in het lichaam. Deze informatie geven zij dan weer door aan de directiekamer. Heel soms maken de procesleiders zelfstandig een beslissing. Deze beslissing kan dan direct worden doorgegeven aan de juiste plaats, waardoor de directiekamer minder werk hoeft te doen. Dit worden ook wel reflexen genoemd. Een bekend voorbeeld is het terugtrekken van je hand wanneer je iets aanraakt wat heet en pijnlijk is.
De zenuwen (perifere zenuwstelsel)
Informatie naar en van alle verschillende plaatsen in het lichaam wordt (in de vorm van elektrische impulsen) vervoerd via miljoenen zenuwen. Zenuwen functioneren dus eigenlijk als de transportbanen van de fabriek. Er zijn transportbanen om informatie vanuit de hersenen (via het ruggenmerg) naar de spieren en organen te vervoeren. Ook zijn er transportbanen om informatie vanuit het lichaam (via het ruggenmerg) naar de hersenen te vervoeren. Om informatie snel en nauwkeurig te vervoeren zit om de zenuwen een vettige isolatielaag, het ‘myeline’. Myeline heeft een witte kleur en wordt daarom ook wel ‘witte stof’ genoemd.
De witte en grijze stof
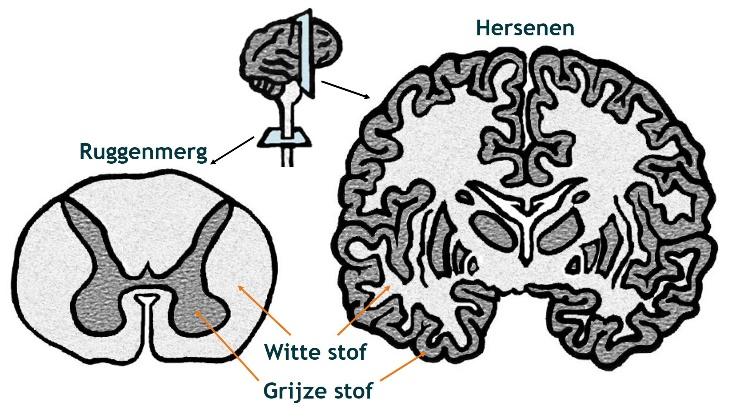 De hersenen en het ruggenmerg bestaan uit witte en grijze stof. In de grijze stof zitten de zenuwcellen of neuronen. In de witte stof zitten de uitlopers van de neuronen die weer verbindingen vormen met andere zenuwcellen in de hersenen of het ruggenmerg. In de witte stof zitten ook andere cellen, bijvoorbeeld oligodendrocyten die beschermende isolatielaagjes voor de dunne zenuwuitlopers vormen (deze laagjes noemen we myeline), maar ook bloedvaatjes, afweercellen en astrocyten.
De hersenen en het ruggenmerg bestaan uit witte en grijze stof. In de grijze stof zitten de zenuwcellen of neuronen. In de witte stof zitten de uitlopers van de neuronen die weer verbindingen vormen met andere zenuwcellen in de hersenen of het ruggenmerg. In de witte stof zitten ook andere cellen, bijvoorbeeld oligodendrocyten die beschermende isolatielaagjes voor de dunne zenuwuitlopers vormen (deze laagjes noemen we myeline), maar ook bloedvaatjes, afweercellen en astrocyten.
In de hersenen bevindt de witte stof zich aan de binnenkant en de grijze stof zich met name aan de buitenkant. In het ruggenmerg zit de witte stof aan de buitenkant en de grijze stof aan de binnenkant.
MLD is een wittestofziekte. Een hersenziekte wordt een wittestofziekte genoemd als primair de witte stof in het centrale zenuwstelsel aangetast is. Bij MLD wordt de witte stof in eerste instantie normaal aangemaakt, maar wordt in de loop van de ziekte geleidelijk afgebroken. Dit noemen we demyelinisatie. Een erfelijke wittestofziekte wordt ook wel een leukodystrofie genoemd. Leukodystrofie is afgeleid van het Griekse woorden ‘leuko’, wat wit betekent, en ‘dystrofie’, wat afbraak betekent.
De oorzaak
Bij MLD wordt demyelinisatie veroorzaakt door een verstoorde stofwisseling van het myeline. Dit komt doordat sulfatide een belangrijke bouwsteen van het myeline is. ASA-deficiëntie leidt tot sulfatidestapeling in het myeline, waardoor het myeline beschadigd en afgebroken wordt. Dit gebeurt niet alleen in het centrale zenuwstelsel. Ook de witte stof rondom de zenuwen raakt beschadigd. Hoe snel demyelinisatie optreedt, is afhankelijk van de snelheid van sulfatidestapeling. Hoe groter het ASA-tekort is, hoe sneller de sulfatidestapeling plaatsvindt, en hoe eerder en sneller demyelinisatie optreedt. Om deze reden maken we onderscheid tussen drie verschillende vormen van MLD:
- laat-infantiele vorm: bij deze vorm is de hoeveelheid ASA zeer laag of zelfs helemaal afwezig. De klachten door demyelinisatie beginnen voor een leeftijd van 2,5 jaar.
- juveniele vorm: bij deze vorm is de hoeveelheid ASA iets hoger dan bij de laat-infantiele vorm. Demyelinisatie en de klachten hiervan verlopen hierdoor trager, waardoor de klachten op een leeftijd tussen de 2,5 en 16 jaar beginnen.
- volwassen (adulte) vorm: bij deze vorm is de hoeveelheid ASA hoger dan bij de andere vormen, maar nog steeds lager dan normaal. De klachten door demyelinisatie beginnen daardoor pas na een leeftijd van 16 jaar.
Erfelijkheid
ASA-deficiëntie is al bij de geboorte aanwezig. Een stofwisselingsziekte is namelijk een erfelijke ziekte. Dit betekent dat de ziekte het gevolg is van een foutje (mutatie) in de genen. Bij MLD zit de mutatie in het ARSA-gen. Dit is het gen dat verantwoordelijk is voor de aanmaak van ASA. Er zijn meer dan 150 mutaties in het ARSA-gen bekend. De verschillende mutaties veroorzaken (deels) hoe ernstig de ASA-deficiëntie is. Hierdoor worden sommige mutaties vaker gezien bij MLD-patiënten met de laat-infantiele vorm, terwijl andere mutaties vaker worden gezien bij MLD-patiënten met de adulte vorm.
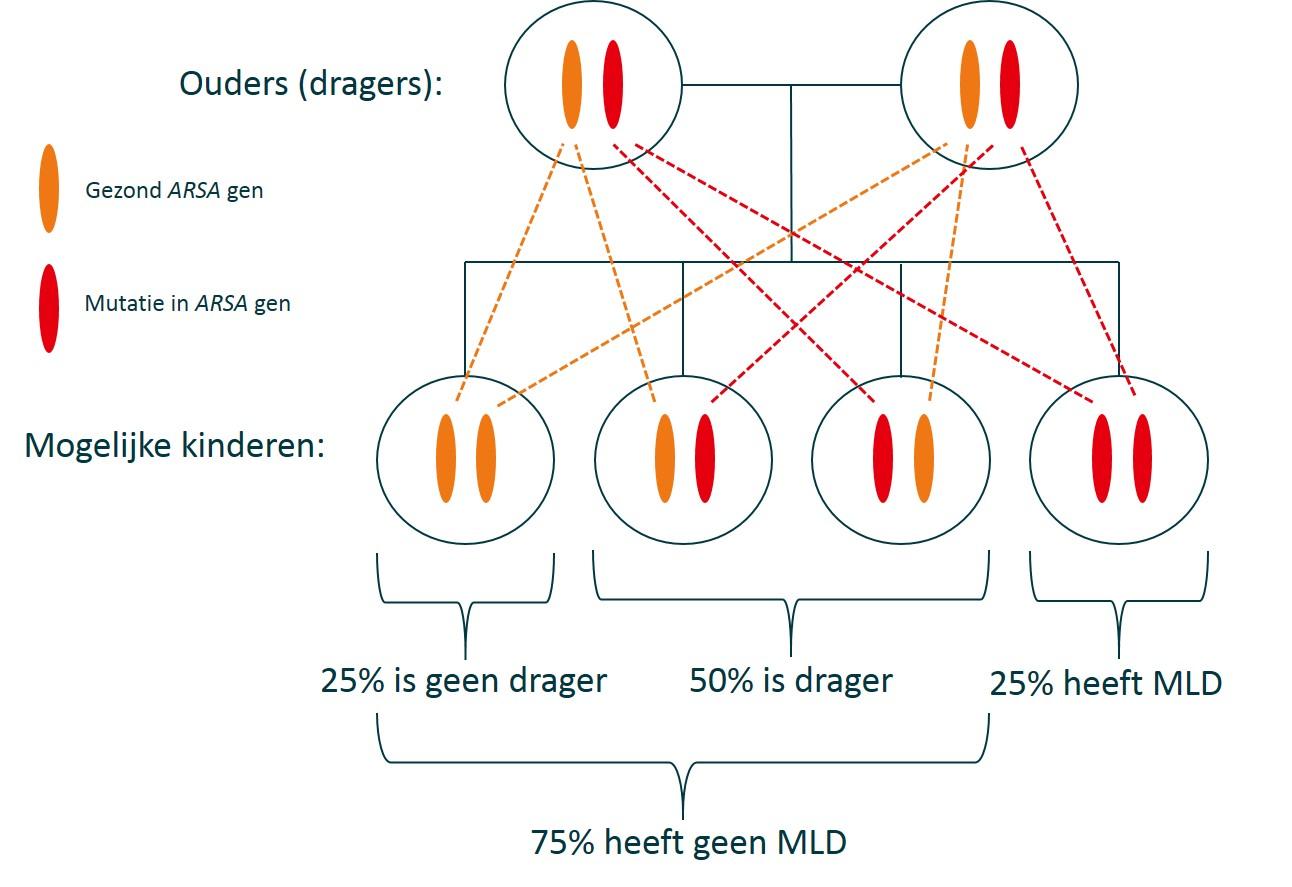
Een gen bestaat uit twee varianten, waarvan één is ‘overgeërfd’ van de moeder en de ander van de vader. Sommige ziektes worden veroorzaakt door een mutatie in één gen-variant (‘dominant’ erfelijke ziektes). MLD wordt veroorzaakt als in beide ARSA-varianten een mutatie aanwezig is. Dit is een ‘recessief’ erfelijke ziekte. Eén persoon kan twee dezelfde (homozygote) of twee verschillende (heterozygote) mutaties in het ARSA-gen hebben. Wanneer iemand een mutatie in slecht één ARSA-variant heeft, heeft die persoon niet de ziekte MLD. Wel wordt die persoon een ‘drager’ van de mutatie genoemd.
Een drager heeft 50% kans om de mutatie door te geven aan zijn of haar kinderen. Wanneer beide ouders drager zijn, heeft een kind 25% kans om van beide ouders de mutatie te krijgen (0,5 (50%) x 0,5 (50%) = 0,25 (25%)). Een broer of zus van iemand met MLD heeft dus 25% kans om ook de ziekte te hebben.
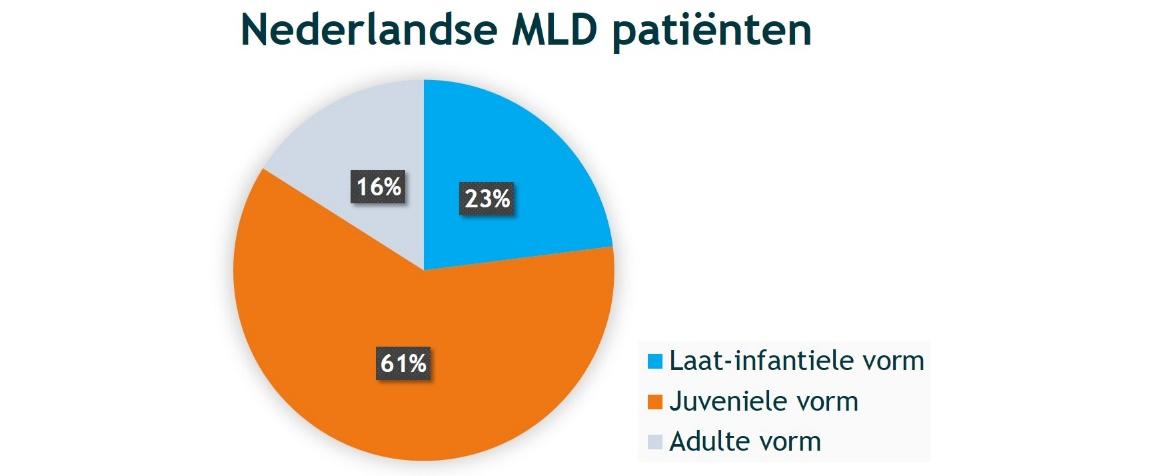 MLD is een zeldzame ziekte. In Nederland wordt de diagnose elk jaar bij ongeveer vijf nieuwe mensen gesteld. Dit betekent dat de ziekte bij ongeveer één op de 70.000 levend geboren kinderen voorkomt. De meeste Nederlandse MLD-patiënten hebben de juveniele vorm, namelijk ongeveer 60%. Van de overige Nederlandse MLD-patiënten heeft ongeveer 25% de laat-infantiele vorm en ongeveer 15% de adulte vorm. Wereldwijd komt de laat-infantiele vorm het meest voor.
MLD is een zeldzame ziekte. In Nederland wordt de diagnose elk jaar bij ongeveer vijf nieuwe mensen gesteld. Dit betekent dat de ziekte bij ongeveer één op de 70.000 levend geboren kinderen voorkomt. De meeste Nederlandse MLD-patiënten hebben de juveniele vorm, namelijk ongeveer 60%. Van de overige Nederlandse MLD-patiënten heeft ongeveer 25% de laat-infantiele vorm en ongeveer 15% de adulte vorm. Wereldwijd komt de laat-infantiele vorm het meest voor.
Doordat de witte stof wel goed wordt aangelegd, hebben MLD-patiënten bij de geboorte geen verschijnselen van de ziekte. Ook de ontwikkeling verloopt in eerste instantie meestal normaal. Als gevolg van de sulfatidestapeling ontstaan in de loop van de tijd de klachten of ziekteverschijnselen. Wanneer deze ontstaan en wat de aard, ernst en het beloop van deze klachten en ziekteverschijnselen zijn, kan verschillen tussen patiënten met zowel een andere als dezelfde MLD-vorm, soms zelfs binnen één gezin.
Laat-infantiele MLD
Bij kinderen met laat-infantiele MLD treden de ziekteverschijnselen voor een leeftijd van 2,5 jaar op. Meestal uiten deze zich als ontwikkelingsproblemen (vertraging, stilstand of achteruitgang van ontwikkeling), vaak van de grove motoriek, zoals lopen. Een aantal kinderen leert nooit lopen, terwijl anderen vaker vallen of het lopen verleren. Ook hebben ze vaak problemen met de fijne motoriek (bijvoorbeeld trillende handen of moeite met oogbewegingen) en problemen met het spreken. Als de ziekteverschijnselen tot uiting komen, verloopt de ziekte meestal snel. De spierkracht neemt af waardoor de kinderen niet meer zelfstandig kunnen staan en zitten. De verstandelijke ontwikkeling stopt en het zien, horen en spreken worden minder. Door problemen met slikken wordt sondevoeding nodig. Na een tijdje worden de spieren te gespannen. Dit heet spasticiteit. Ook ontstaat er bij 25 tot 50% van de kinderen epilepsie. De kinderen zijn binnen een paar jaar bedlegerig en volledig zorgafhankelijk. In eerste instantie reageren ze nog op hun ouders, maar uiteindelijk verliezen ze door het contact met hun omgeving. De meeste kinderen overlijden tussen 2 en 7 jaar na het optreden van de eerste ziekteverschijnselen.
Juveniele MLD
Bij kinderen met juveniele MLD ontstaan de eerste ziekteverschijnselen tussen hun derde en zestiende levensjaar. Dit kunnen problemen in de grove motoriek zijn, maar veel vaker valt een achteruitgang in schoolprestaties of gedragsverandering op. Doordat deze problemen ook andere oorzaken kunnen hebben, zoals ADHD, wordt de diagnose meestal later gesteld dan bij kinderen met laat-infantiele MLD. De diagnose komt vaak aan het licht als er ook lichamelijke verschijnselen optreden, zoals een toenemende onhandigheid, vreemd lopen of slordiger praten. Net als kinderen met laat-infantiele MLD, verliezen deze kinderen in de loop van tijd hun lichamelijke en verstandelijke vaardigheden, ontstaan er slikproblemen en spasticiteit en krijgt 50 tot 60% epilepsie. Het ziekteverloop is veelal langzamer, maar door bijkomende complicaties, zoals een longontsteking, kan de ziekte onverwachts versneld worden. De levensverwachting is hierdoor moeilijk in te schatten en varieert tussen de 3 en 15 jaar na het optreden van de eerste ziekteverschijnselen.
Adulte MLD
Bij patiënten met adulte MLD ontstaan de eerste ziekteverschijnselen vaak na de puberteit, maar de precieze leeftijd kan sterk variëren. Bij de meeste patiënten begint de ziekte met gedragsproblemen, persoonlijkheidsveranderingen, geheugenproblemen of psychiatrische verschijnselen. Hierdoor wordt vaak eerst gedacht aan een burn-out, depressie of andere psychiatrische ziekte. De lichamelijke verschijnselen kunnen tot jaren na begin van de ziekte pas optreden. Bij een klein deel van de patiënten treden de lichamelijke verschijnselen juist als eerste op. De oorzaak hiervan is nog niet bekend. Wel is bekend dat het ziekteverloop langzamer is dan bij laat-infantiele en juveniele MLD. Daarnaast hebben deze patiënten minder vaak epilepsie. Uiteindelijk gaan ook bij deze vorm alle lichamelijke en verstandelijke vaardigheden verloren, ontstaan er slikproblemen en spasticiteit, en worden patiënten volledig zorgafhankelijk. De levensverwachting varieert tussen de 5 en 35 jaar na het optreden van de eerste ziekteverschijnselen.
Dit is een overzicht van de meest voorkomende klachten en ziekteverschijnselen. De klachten en ziekteverschijnselen hoeven niet allemaal tegelijk aanwezig te zijn en kunnen veranderen in de tijd. Indien u hierover vragen heeft, stel deze dan gerust aan de arts.
Problemen met grove en/of fijne motoriek |
Concentratie- en aandachtsproblemen |
Spierzwakte |
Geheugenproblemen |
(Pijnlijke) spasticiteit (stijve spieren) |
Achteruitgang leerprestaties |
Verlies van lichamelijke vaardigheden |
Verandering van gedrag of persoonlijkheid |
Onbedoelde bewegingen |
Onduidelijk praten of verlies van spraak |
Balans- en coördinatieproblemen |
Problemen met slikken en ondervoeding |
Verminderd gevoel bij aanraking |
Problemen met zien en horen |
Pijnlijk gevoel bij aanraking |
Epileptische aanvallen |
Afwijkende stand van de voeten |
Galblaasproblemen, mogelijk met buikpijn |
Problemen met plassen |
Problemen met de ontlasting |
De diagnose wordt meestal gesteld door een neuroloog. De neuroloog luistert naar het verhaal van de patiënt en op basis daarvan stelt hij voor om aanvullende onderzoeken te doen. Eén van de meest gebruikte onderzoeken is een MRI-scan van de hersenen.
MRI-scan (magnetic resonance imaging)
De afkorting MRI staat voor magnetic resonance imaging (beeldvorming met magnetische resonantie). Bij een MRI-scan worden signalen in het lichaam opgewekt met behulp van een grote sterke magneet en radiogolven. Deze signalen worden vervolgens door een antenne opgevangen en door een computer verwerkt tot afbeeldingen. Er wordt dus geen röntgenstraling gebruikt. De afbeeldingen tonen doorsnedes van de hersenen op verschillende locaties, alsof er plakjes van de hersenen zijn gesneden.
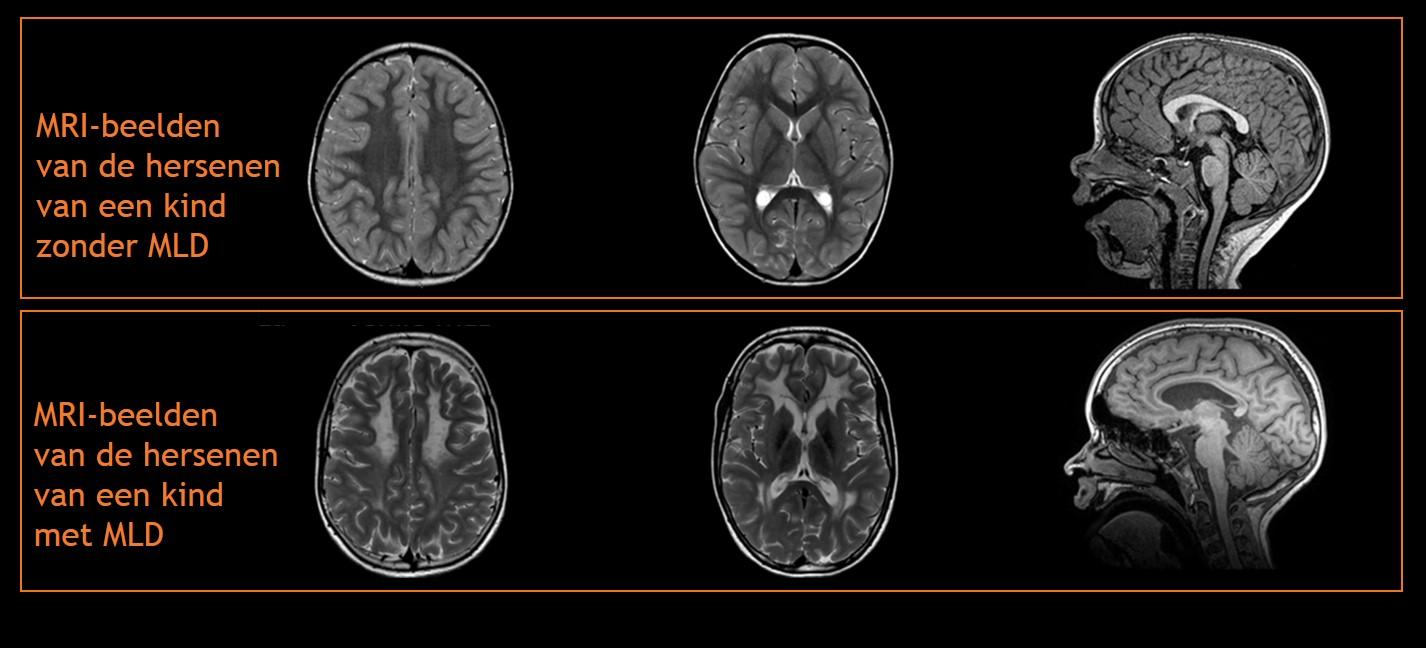 Op de MRI-afbeeldingen kan de neuroloog afwijkingen in de witte stof zien die goed passen bij de ziekte MLD. In de beginfase van de ziekte, als er nog geen of weinig ziekteverschijnselen zijn, kunnen deze afwijkingen nog afwezig of slechts heel subtiel zijn. Naar mate de ziekte zich vordert, breiden de afwijkingen zich uit op meerdere plekken in de hersenen. Hieronder ziet u een aantal MRI-afbeeldingen van de hersenen van een kind zonder MLD en van de hersenen van een kind met MLD. De MRI-afbeeldingen van het tweede kind zijn vijf jaar na het ontstaan van de eerste ziekteverschijnselen gemaakt.
Op de MRI-afbeeldingen kan de neuroloog afwijkingen in de witte stof zien die goed passen bij de ziekte MLD. In de beginfase van de ziekte, als er nog geen of weinig ziekteverschijnselen zijn, kunnen deze afwijkingen nog afwezig of slechts heel subtiel zijn. Naar mate de ziekte zich vordert, breiden de afwijkingen zich uit op meerdere plekken in de hersenen. Hieronder ziet u een aantal MRI-afbeeldingen van de hersenen van een kind zonder MLD en van de hersenen van een kind met MLD. De MRI-afbeeldingen van het tweede kind zijn vijf jaar na het ontstaan van de eerste ziekteverschijnselen gemaakt.
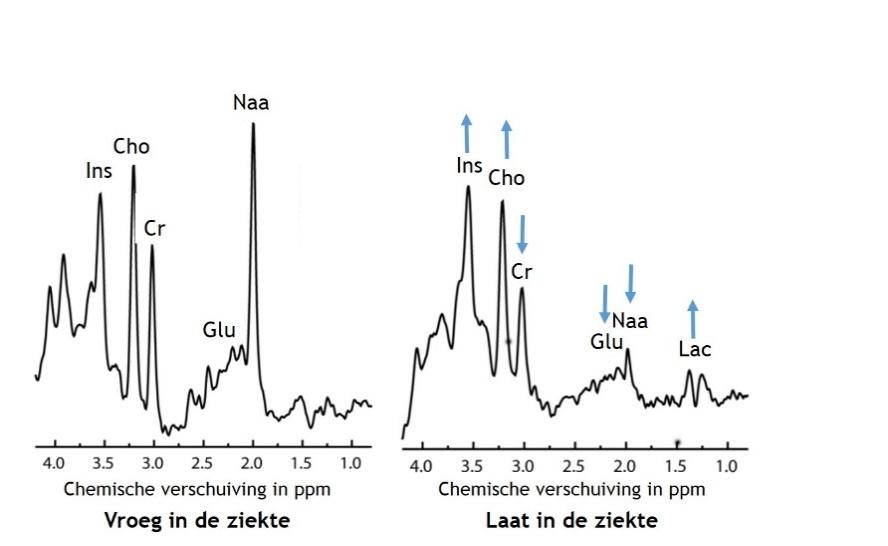 MR-spectroscopie
MR-spectroscopie
Soms wordt er naast het maken van ‘normale’ MRI-afbeeldingen ook een MR-spectroscopie gemaakt. Met een MR-spectroscopie wordt gekeken of bepaalde stofjes in de hersenen aanwezig zijn en of ze verhoogd of verlaagd zijn. Hiermee kan het effect van de ziekte op de hersenen nog beter worden geanalyseerd.
EMG (elektromyografie)
 Wanneer de neuroloog denkt dat er (ook) afwijkingen aan de zenuwen zijn, zal een EMG-onderzoek worden gedaan. De afkorting EMG staat voor elektromyografie. Met het EMG-onderzoek wordt de werking van de zenuwen en de spieren gemeten. Het onderzoek bestaat meestal uit twee delen. Voor het eerste deel worden een aantal elektroden (meestal metalen dopjes) op de huid geplaatst. Deze elektroden zijn met een draad verbonden met de EMG-computer. De zenuw wordt vervolgens door andere elektroden op de huid geprikkeld door middel van korte schokjes. Deze schokjes duren een fractie van een seconde, waarbij er steeds een stootje wordt gevoeld. Op de monitor van de EMG-computer is te zien hoe de zenuwen reageren op deze schokjes. Dit deel van het EMG-onderzoek wordt ook wel geleidingsonderzoek genoemd.
Wanneer de neuroloog denkt dat er (ook) afwijkingen aan de zenuwen zijn, zal een EMG-onderzoek worden gedaan. De afkorting EMG staat voor elektromyografie. Met het EMG-onderzoek wordt de werking van de zenuwen en de spieren gemeten. Het onderzoek bestaat meestal uit twee delen. Voor het eerste deel worden een aantal elektroden (meestal metalen dopjes) op de huid geplaatst. Deze elektroden zijn met een draad verbonden met de EMG-computer. De zenuw wordt vervolgens door andere elektroden op de huid geprikkeld door middel van korte schokjes. Deze schokjes duren een fractie van een seconde, waarbij er steeds een stootje wordt gevoeld. Op de monitor van de EMG-computer is te zien hoe de zenuwen reageren op deze schokjes. Dit deel van het EMG-onderzoek wordt ook wel geleidingsonderzoek genoemd.
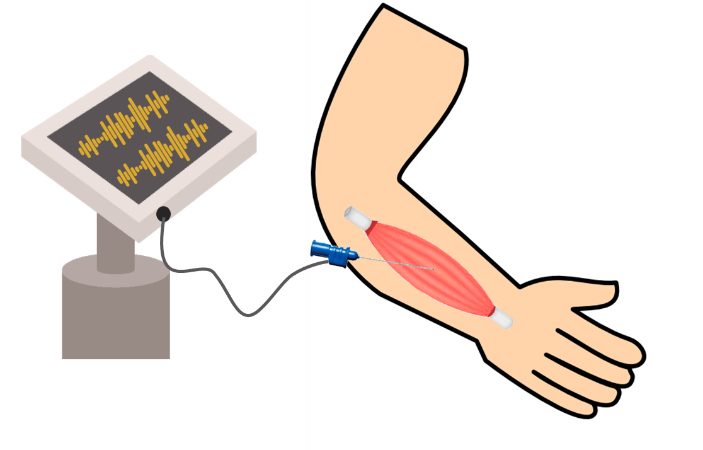 Voor het tweede deel wordt een naaldje in de spier geprikt. De arts beweegt vervolgens het naaldje in de spier. Dit kan wel wat pijnlijk zijn. Het naaldje is net als de elektroden via een draad verbonden met de EMG-computer. De activiteit van de aangeprikte spier wordt op de monitor van het EMG-computer geregistreerd. U kunt deze activiteit zelf ook horen. Dit deel van het EMG-onderzoek wordt ook wel naaldonderzoek genoemd.
Voor het tweede deel wordt een naaldje in de spier geprikt. De arts beweegt vervolgens het naaldje in de spier. Dit kan wel wat pijnlijk zijn. Het naaldje is net als de elektroden via een draad verbonden met de EMG-computer. De activiteit van de aangeprikte spier wordt op de monitor van het EMG-computer geregistreerd. U kunt deze activiteit zelf ook horen. Dit deel van het EMG-onderzoek wordt ook wel naaldonderzoek genoemd.
Urineonderzoek
Als de neuroloog op basis van de ziekteverschijnselen en de bevindingen bij MRI-onderzoek en/of EMG-onderzoek denkt aan de ziekte MLD, kan met behulp van urineonderzoek de diagnose worden vastgesteld of uitgesloten. Het overschot aan sulfatides in het lichaam veroorzaakt namelijk sulfatidestapeling in de nieren en de urine. Bij urineonderzoek van MLD-patiënten wordt dan ook altijd een verhoogde concentratie sulfatides gevonden.
Bloedonderzoek
Naast het urineonderzoek wordt het bloed onderzocht. In het bloed kan de activiteit van het enzym ASA gemeten worden. Bij de ziekte MLD is deze activiteit altijd verlaagd. Sommige mensen hebben wel een verlaagde activiteit van ASA, maar niet een verhoogde concentratie sulfatides in de urine. In de medische wereld wordt dit fenomeen ‘pseudo-arylsulfatase A deficiëntie’ genoemd. Deze mensen hebben niet de ziekte MLD en krijgen ook geen ziekteverschijnselen. Pseudo-arylsulfatase A deficiëntie wordt ook veroorzaakt door varianten in het ARSA-gen, maar dit zijn andere varianten dan de mutaties die MLD veroorzaken. Een aantal patiënten hebben zowel de mutaties die MLD veroorzaken als de mutaties die pseudo-arylsulfatase A deficiëntie veroorzaken.
Genetisch onderzoek (DNA-onderzoek) en familieonderzoek
In het bloed kan ook het ARSA-gen worden onderzocht. Dit wordt genetisch onderzoek of DNA-onderzoek genoemd. Met dit onderzoek wordt bepaald welke twee mutaties in het ARSA-gen de ziekte veroorzaken. Dit is van belang om de diagnose compleet te maken en om eventueel een familieonderzoek uit te voeren. Een familieonderzoek wordt gedaan om te achterhalen of familieleden van de patiënt, zoals een broer, zus of kind, ook de ziekte hebben of drager zijn van de ziekte (lees voor meer informatie sectie 3: erfelijkheid). Met behulp van familieonderzoek kan de diagnose worden gesteld voordat iemand klachten of ziekteverschijnselen heeft. Dit is van belang voor de behandeling van MLD.
Prenatale diagnostiek en preïmplantatie genetische diagnostiek
Familieonderzoek kan ook zinvol zijn voor een toekomstige zwangerschap en de keuze voor prenatale diagnostiek of preïmplantatie genetische diagnostiek. Met prenatale diagnostiek wordt tijdens de zwangerschap door middel van een vlokkentest of vruchtwaterpunctie onderzocht of de baby MLD heeft. Meer informatie hierover kunt u vinden op www.onderzoekvanmijnongeborenkind.nl. Bij preïmplantatie genetische diagnostiek (PGD) vindt de bevruchting van meerdere eicellen (door zaadcellen) buiten het lichaam plaats met behulp van IVF (In Vitro Fertilisatie). Vervolgens wordt met genetisch onderzoek onderzocht of de bevruchte eicellen (embryo’s) de ziekte MLD hebben. Daarna worden alleen embryo’s die geen MLD hebben, teruggeplaatst in de baarmoeder. Hierover kunt u meer lezen op de website van PGD Nederland www.pgdnederland.nl.
 Welke onderzoeken zijn er nog meer nodig?
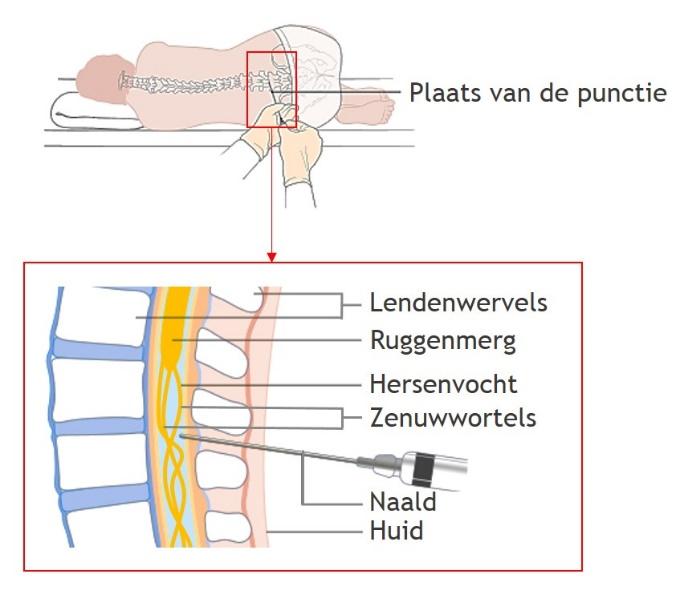
Welke onderzoeken zijn er nog meer nodig?Onderzoek van het hersenvocht (liquor)
 De hersenen en het ruggenmerg worden omgeven door een waterige vloeistof, het hersenvocht (liquor). Het hersenvocht is belangrijk voor de bescherming van de hersenen en het ruggenmerg. Daarnaast zorgt het hersenvocht voor het transport van voedingsstoffen de afvoer van afvalstoffen. De aanwezigheid en concentraties van verschillende stofjes in het hersenvocht kunnen daardoor wat zeggen over de conditie en gezondheid van de hersenen. Om deze reden wordt onderzoek van het hersenvocht verricht. Dit gebeurt door middel van een ruggenprik, ook wel lumbaalpunctie genoemd. Hierbij prikt de neuroloog met een dunne naald tussen twee wervels in de onderrug en wordt er circa 5ml hersenvocht afgenomen. Deze hoeveelheid hersenvocht kan zonder problemen worden afgenomen, omdat er elke dag 500ml hersenvocht door het lichaam wordt aangemaakt. Desondanks kunnen mensen na de lumbaalpunctie hoofdpijn ervaren door een (tijdelijk) verlaagde druk van het hersenvocht.
De hersenen en het ruggenmerg worden omgeven door een waterige vloeistof, het hersenvocht (liquor). Het hersenvocht is belangrijk voor de bescherming van de hersenen en het ruggenmerg. Daarnaast zorgt het hersenvocht voor het transport van voedingsstoffen de afvoer van afvalstoffen. De aanwezigheid en concentraties van verschillende stofjes in het hersenvocht kunnen daardoor wat zeggen over de conditie en gezondheid van de hersenen. Om deze reden wordt onderzoek van het hersenvocht verricht. Dit gebeurt door middel van een ruggenprik, ook wel lumbaalpunctie genoemd. Hierbij prikt de neuroloog met een dunne naald tussen twee wervels in de onderrug en wordt er circa 5ml hersenvocht afgenomen. Deze hoeveelheid hersenvocht kan zonder problemen worden afgenomen, omdat er elke dag 500ml hersenvocht door het lichaam wordt aangemaakt. Desondanks kunnen mensen na de lumbaalpunctie hoofdpijn ervaren door een (tijdelijk) verlaagde druk van het hersenvocht.
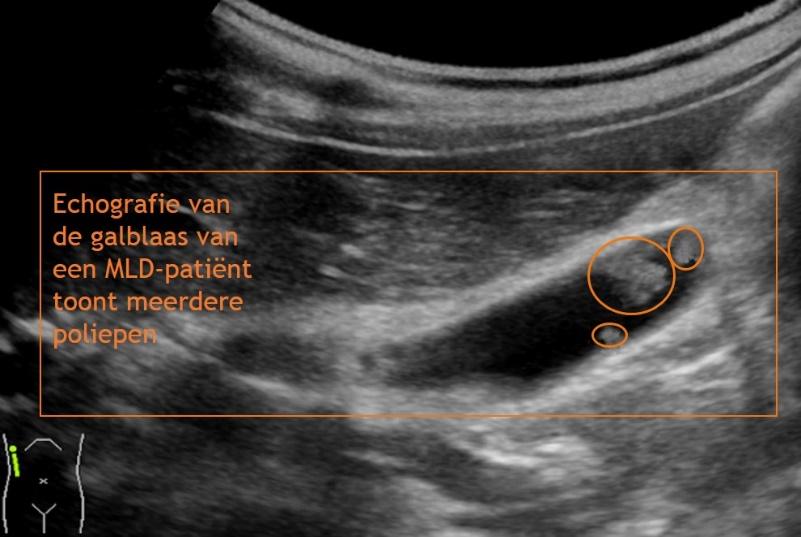
Echografie van de galblaas
MLD-patiënten hebben een grotere kans op het ontwikkelen van galstenen, een ontsteking van de galblaas of poliepen in de galblaas. Dit kan leiden tot buikpijnklachten, meestal rechts in de bovenbuik. Daarnaast geeft dit een hogere kans op galblaaskanker. Met behulp van echografie wordt gekeken of er sprake is van één of meerdere van deze galblaasproblemen. Indien dit het geval is, wordt geadviseerd de galblaas door middel van een kijkoperatie te verwijderen.
Neuropsychologisch onderzoek
Het neuropsychologisch onderzoek wordt verricht door een neuropsycholoog. Het bestaat uit een gesprek, verschillende testen (waaronder kleine puzzeltjes) en soms ook vragenlijsten. De neuropsycholoog kijkt gedurende het neuropsychologisch onderzoek naar het cognitief functioneren (zoals aandacht, geheugen, taal, denken en het uitvoeren van handelingen), het gedrag en emoties. Daarnaast wordt een intelligentie-onderzoek gedaan.
EEG (elektro-encefalografie)
Als iemand epileptische aanvallen krijgt, zal een hersenfilmpje worden gemaakt. In medische termen wordt dit een EEG genoemd, wat staat voor elektro-encefalografie. Tijdens het onderzoek wordt met behulp van elektroden op de hoofdhuid de elektrische activiteit (hersengolven) van de hersenen gemeten. De elektroden zijn via draadjes verbonden met een computer. De hersengolven worden op deze manier aan de computer doorgegeven. Bij normale hersenactiviteit wisselen snelle en langzamere hersengolven elkaar af. Dit is zichtbaar op het EEG op het computerscherm. Tijdens een epileptische aanval is de activiteit van de hersenen verstoord. Op het computerscherm is dan een andere activiteit te zien.
Onderzoek van de ogen
Als gevolg van MLD wordt de oogzenuw dun. Dit wordt ook wel opticusatrofie genoemd. Dit kan leiden tot wazig zien, uitval van het gezichtsveld, verminderd contrast of kleuren zien en afwijkende reacties van de pupil. Daarom worden patiënten vaak onderzocht door de oogarts. De oogarts onderzoekt de visus, het gezichtsveld en hoe de oogzenuw er uitziet.
Op dit moment is MLD niet te genezen. Er is ook nog geen behandeling om alle gevolgen van MLD te voorkomen. Bij een deel van patiënten wordt een hematopoëtische stamceltransplantatie (ook wel bekend als beenmergtransplantatie) toegepast om het lichaam voldoende ASA te laten maken vanuit de nieuwe donorcellen. Het duurt ongeveer één jaar voordat het lichaam dit kan en deze behandeling effectief is. Het is daarom niet zinvol om deze behandeling toe te passen bij MLD-patiënten met de laat-infantiele vorm of bij MLD-patiënten met al duidelijke ziekteverschijnselen. Bij deze MLD-patiënten kan de behandeling het beloop van de ziekte juist ongewenst versnellen. Ook is de behandeling niet bij alle patiënten werkzaam. In sommige gevallen komen patiënten in aanmerking voor een experimentele behandeling. Indien dit niet het geval is, worden de klachten en ziekteverschijnselen van de patiënt zo goed mogelijk behandeld. Er is helaas nog geen medicijn bekend dat de ziekte kan vertragen.
-
- Hematopoëtische stamceltransplantatie (beenmergtransplantatie)
Bij patiënten die vroeg of zelfs voor het begin van de ziekteverschijnselen (bijvoorbeeld door familieonderzoek) met MLD gediagnosticeerd worden, is een hematopoëtische stamceltransplantatie de aangewezen behandeling. Voordat de behandeling plaatsvindt, worden patiënten uitgebreid onderzocht. Op basis van een lichamelijk onderzoek, het neuropsychologisch onderzoek en de MRI- en MR-spectroscopie uitkomsten wordt besloten of een stamceltransplantatie nog mogelijk is. Als dit het geval is, wordt de patiënt doorverwezen naar een transplantatiecentrum. In Nederland is dit het UMC Utrecht. Daar wordt vervolgens onderzocht of de patiënt de stamceltransplantatie aankan en of er een geschikte donor beschikbaar is.
Wat doen stamcellen?
Het lichaam bestaat uit veel verschillende soorten cellen met eigen functies. Zo bestaan de hersenen uit andere soorten cellen dan de lever. Hersencellen kunnen ook niet ontwikkelen tot levercellen of andersom. De unieke kenmerken van stamcellen zijn dat zij zich wel tot verschillende soorten cellen kunnen ontwikkelen en zich ook blijven vermenigvuldigen in het lichaam. De stamcellen die worden gebruikt in een stamceltransplantatie kunnen zich ontwikkelen tot verschillende soorten bloedcellen. Voor MLD is een bepaald soort witte bloedcellen (afweercellen), de macrofagen, van belang. De macrofagen (afkomstig van een donor) zijn namelijk cellen die ASA kunnen produceren en afgeven in het lichaam van de patiënt, bijvoorbeeld in het zenuwstelsel. Daarnaast kunnen de donormacrofagen met behulp van ASA de sulfatides in het lichaam opruimen.
Wat houdt de stamceltransplantatie in?
 Voordat een stamceltransplantatie uitgevoerd kan worden, moet er in het lichaam van de patiënt ruimte zijn voor de donorcellen. Anders kunnen de donorcellen worden afgestoten. Daarom worden de stamcellen en de afweercellen van de patiënt eerst vernietigd met behulp van medicijnen, waaronder intensieve chemotherapie. Dit wordt ook wel ‘conditionering’ genoemd en dit duurt ongeveer één week. Daarna vindt de stamceltransplantatie plaats met stamcellen afkomstig uit beenmerg of navelstrengbloed van een donor. De stamcellen worden via een infuus aan het bloed van de patiënt gegeven. Vervolgens reizen ze via het bloed naar het beenmerg. Daar ontwikkelen de stamcellen zich tot nieuwe bloed-, afweer- en beenmergcellen. Dit duurt ongeveer drie weken. Deze periode heet ‘de dip’, omdat de patiënt tijdens deze periode extra gevoelig voor infecties is. De stamcellen reizen via het bloed ook naar de hersenen. Daar ontwikkelen ze zich tot de macrofagen die ASA produceren en sulfatides opruimen. Het duurt ongeveer één jaar voordat er voldoende ASA producerende macrofagen in de hersenen zijn om de ziekte tot stilstand te brengen.
Voordat een stamceltransplantatie uitgevoerd kan worden, moet er in het lichaam van de patiënt ruimte zijn voor de donorcellen. Anders kunnen de donorcellen worden afgestoten. Daarom worden de stamcellen en de afweercellen van de patiënt eerst vernietigd met behulp van medicijnen, waaronder intensieve chemotherapie. Dit wordt ook wel ‘conditionering’ genoemd en dit duurt ongeveer één week. Daarna vindt de stamceltransplantatie plaats met stamcellen afkomstig uit beenmerg of navelstrengbloed van een donor. De stamcellen worden via een infuus aan het bloed van de patiënt gegeven. Vervolgens reizen ze via het bloed naar het beenmerg. Daar ontwikkelen de stamcellen zich tot nieuwe bloed-, afweer- en beenmergcellen. Dit duurt ongeveer drie weken. Deze periode heet ‘de dip’, omdat de patiënt tijdens deze periode extra gevoelig voor infecties is. De stamcellen reizen via het bloed ook naar de hersenen. Daar ontwikkelen ze zich tot de macrofagen die ASA produceren en sulfatides opruimen. Het duurt ongeveer één jaar voordat er voldoende ASA producerende macrofagen in de hersenen zijn om de ziekte tot stilstand te brengen.
Afweerreacties (Graft-Versus-Host-Disease (GVHD)/transplantatie ziekte)
Een stamceltransplantatie met stamcellen van een donor wordt een allogene stamceltransplantatie genoemd. De donor is een verwant (familielid) of niet-verwant persoon die met de patiënt ‘gematcht’ is aan de hand van een (bijna) identieke weefseltypering (HLA-typering). Dit is belangrijk, omdat de afweercellen van de donor afweerreacties kunnen veroorzaken. Hierbij worden gezonde lichaamscellen van de patiënt aangevallen door de afweercellen van de donor. Dit wordt Graft-Versus-Host-Disease (GVHD) of transplantatieziekte genoemd. De kans op GVHD is altijd aanwezig, ook wanneer de HLA-typering identiek is. Dit komt door andere kleine verschillen tussen de donor en de patiënt. De verschijnselen van GVHD kunnen licht tot ernstig zijn en een korte tijd tot meerdere jaren aanhouden. Daarnaast kan er sprake zijn van een acute en een chronische GVDH vorm.
Acute GVHD treedt binnen de eerste drie maanden na de stamceltransplantatie op. Hierbij kunnen e volgende klachten optreden: koorts, roodheid van de huid, een droge of pijnlijke mond, buikpijn, diarree, braken, geelzucht, droge ogen en hoesten. De behandeling is afhankelijk van de ernst. Een veelgebruikt medicijn is prednison. Deze behandeling is vaak effectief, maar niet altijd. Acute GVHD kan dan overgaan in chronische GVHD.
Chronische GVHD ontstaat vanaf drie maanden na de stamtransplantatie, met of zonder voorafgaande acute GVHD. De meest voorkomende klachten hiervan zijn droge ogen, een droge mond en kleurveranderingen van de huid. In ernstige gevallen kan verstijving van de huid optreden of kan de werking van de lever of longen worden aangetast. Chronische GVHD kan jaren duren. De behandeling bestaat uit medicijnen die de afweercellen onderdrukken, zoals corticosteroïdcrème, prednison en ciclosporine.
Naar huis, nazorg en controles
Patiënten hebben na de stamceltransplantatie, ook na ‘de dip’, tijdelijk een verminderde afweer. Dit komt door de conditionering en het gebruik van afweer onderdrukkende medicatie na de stamceltransplantatie. Daarom is het belangrijk dat de patiënt en zijn of haar omgeving zich aan een aantal leefregels houden. Dit zijn beschermende maatregelen op het gebied van hygiëne, lichaamsverzorging en voeding. De leefregels gelden tijdens de ziekenhuisopname en de eerste zes maanden thuis. Als de patiënt daarna nog afweer onderdrukkende medicatie gebruikt, wordt geadviseerd om deze leefregels te handhaven tot hij of zij deze medicatie niet meer gebruikt. De artsen en verpleegkundigen in het transplantatiecentrum zullen hier meer informatie over geven.
De afweer wordt tevens gecontroleerd door een arts in het transplantatiecentrum. Tijdens deze controle-afspraken wordt ook gekeken naar het effect van de stamceltransplantatie op de ziekte. Voor kinderen gebeurt dit in het speciaal voor kinderen opgerichte Sylvia Tóth Centrum. Ten slotte wordt de patiënt minimaal één keer per jaar gezien door de neuroloog in het Amsterdam Leukodystrofie Centrum. Er wordt dan ook een MRI en MR-spectroscopie van de hersenen gemaakt zodat het effect van de behandeling op de witte stof nauwkeurig kan worden beoordeeld, en een zenuwonderzoek (EMG). Soms is er extra onderzoek nodig, bijvoorbeeld van het bloed of het hersenvocht.
-
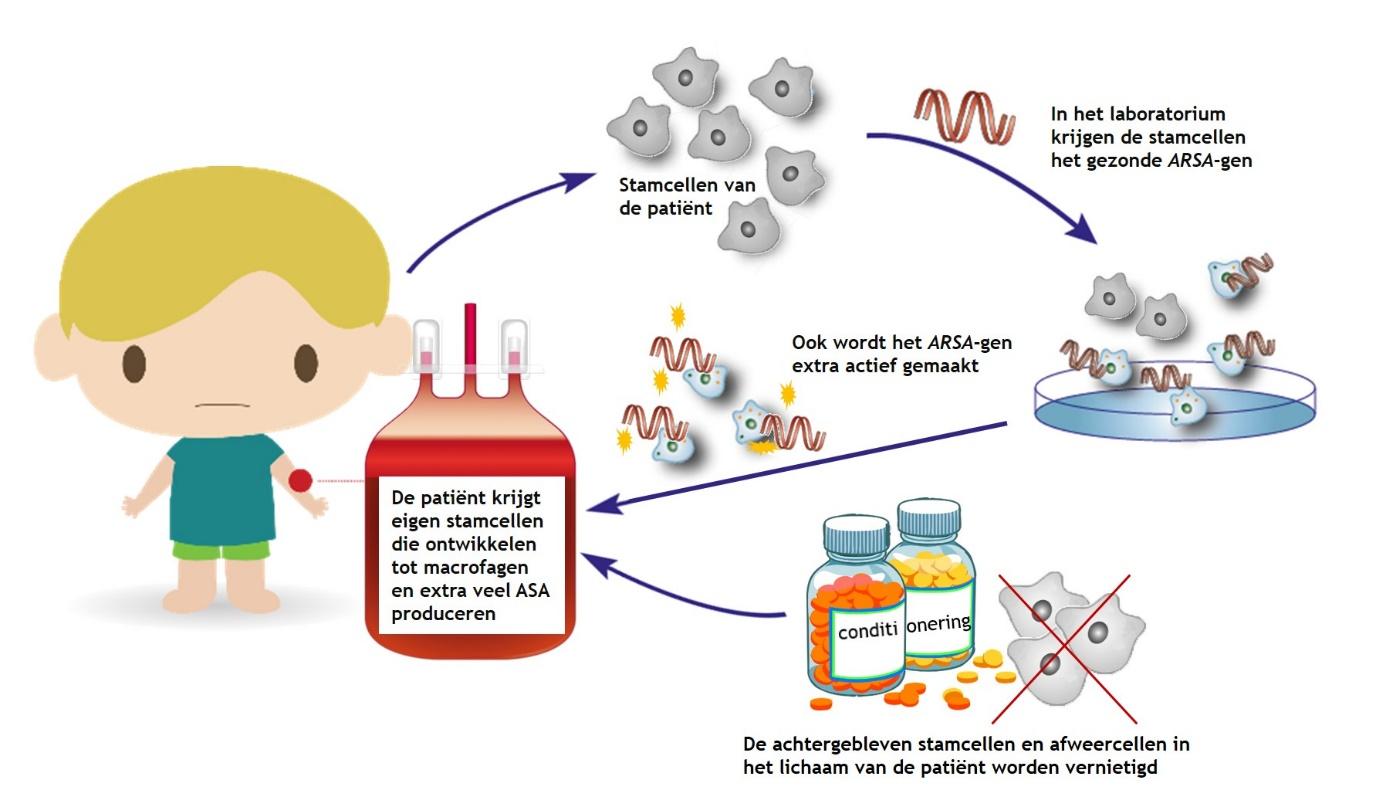 Gentherapie
Gentherapie
Gentherapie voor MLD werkt grotendeels volgens hetzelfde principe als een stamceltransplantatie. Een belangrijk verschil is dat de stamcellen van de patiënt worden gebruikt in plaats van stamcellen van een donor. In het laboratorium wordt het defecte ARSA-gen in de stamcellen van de patiënt vervangen door een gezond ARSA-gen. Vervolgens worden, na de conditionering met medicatie en/of bestraling, de stamcellen met het gezonde ARSA-gen teruggegeven aan de patiënt. In het lichaam van de patiënt ontwikkelen ze zich tot weer tot de macrofagen die (door het gezonde ARSA-gen) ASA produceren en sulfatides opruimen. Een voordeel van deze techniek is dat eigen stamcellen geen afweerreacties veroorzaken. Het gebruik van afweer onderdrukkende medicatie is hierdoor niet nodig. Daarnaast kan het gezonde ARSA-gen extra actief worden gemaakt, waardoor de macrofagen een grotere hoeveelheid ASA dan normaal gaan produceren. Hierdoor kunnen de macrofagen sneller en meer sulfatides opruimen. Op dit moment lijkt gentherapie effectief, al is nog niet bekend hoe het effect op lange termijn is. Gentherapie (Libmeldy®) komt binnenkort beschikbaar voor een
-
- Enzymvervangende therapie
Een andere behandeling die wordt onderzocht is ‘enzym vervangende therapie’ (ERT). Bij deze behandeling wordt het ASA enzym rechtstreeks gegeven aan de patiënt. Uit onderzoek is gebleken dat het geven van ASA via het bloed de ziekte niet stopt of vertraagt. Dit komt doordat het ASA enzym, in tegenstelling tot stamcellen, niet vanuit het bloed naar de hersenen kan reizen. Het enzym wordt tegengehouden door de zogenoemde ‘bloed-hersenbarrière’. Daarom wordt nu in een klinische studie onderzocht of het geven van ASA via het hersenvocht de ziekte wel kan vertragen. Via het hersenvocht wordt de bloed-hersenbarrière ontweken en kan het ASA enzym wel naar de hersenen reizen. Op dit moment komen alleen patiënten met laat-infantiele MLD voor deze experimentele behandeling in aanmerking.
-
- Zorg voor comfort en ondersteuning
Als een stamceltransplantatie of experimentele behandeling niet mogelijk of zinvol is, zijn er geen andere manieren om de ziekte MLD te vertragen. Wel worden de klachten en ziekteverschijnselen zo goed mogelijk behandeld. Hierbij richt de behandeling zich op het zorgen voor comfort en ondersteuning. U komt hiervoor met verschillende zorgverleners in contact, zoals een revalidatiearts, fysiotherapeut, kinderarts, internist of thuiszorg. Samen wordt steeds opnieuw gekeken naar wat u of uw kind op dat moment nodig heeft. In het begin van de ziekte kunnen dit bijvoorbeeld fysiotherapie, hulpmiddelen of woningvoorzieningen zijn. In een later stadium van de ziekte kan het gaan om bijvoorbeeld het behandelen van epilepsie, voeding via een PEG-sonde of behandeling van spasticiteit. De laatste twee behandelingen zullen hieronder kort worden toegelicht. Wij hechten veel waarde aan de gedachten van ouders (in geval van kinderen) of de partner/andere familieleden over wat nog wel een zinvolle behandeling is en wat niet meer. Het bespreken van deze gedachten met elkaar kan de zorg verbeteren. Het belang van de patiënt staat hierbij voorop.
PEG-sonde en Mic-Key button
MLD veroorzaakt problemen met slikken. Om ondervoeding te voorkomen, kan sondevoeding worden gegeven. Sondevoeding is vloeibaar eten dat via een slangetje rechtstreeks naar de maag gaat. Dit gebeurt meestal via een PEG-sonde. Een PEG-sonde bestaat uit een slangetje dat vanuit de maag door de buikwand naar buiten komt. Een aansluitstukje op de buikhuid houdt het slangetje op zijn plek. Aan het uiteinde van het slangetje zit een toedieningssysteem, waardoor sondevoeding en medicatie kan worden gegeven. Na drie maanden kan de PEG-sonde worden vervangen door een Mic-Key button. Het grootste voordeel van een Mic-Key button is dat er geen slangetje meer aan buitenkant van de buik zit. Als u vragen heeft over de PEG-sonde en Mic-Key button kunt u contact opnemen met één van de kinderneurologen van het Amsterdam Leukodystrofie Centrum.
Behandeling van spasticiteit (Baclofenpomp)
De wittestofschade in de hersenen als gevolg van MLD verstoort de signalen die tussen het zenuwstelsel en de spieren worden uitgewisseld. Deze verstoring veroorzaakt een te hoge activiteit in de spieren (spasticiteit). Als gevolg hiervan zijn de spieren heel stijf waardoor bewegen, vooral van de armen en benen, moeilijk gaat. Daarnaast kunnen er oncontroleerbare bewegingen en pijn ontstaan (spasmen). De spasticiteit bij MLD-patiënten kan worden behandeld door middel van een speciale medicijnenpomp, de Baclofenpomp. Dit wordt ook wel Intrathecale Baclofen (ITB) behandeling genoemd. Het medicijn Baclofen vermindert de spasmefrequentie, waardoor de pijn afneemt. Daarnaast verlaagt het de spierspanning, waardoor bewegingen en de zorg wordt vergemakkelijkt en er een kleinere kans is op blijvende standsafwijkingen van de gewrichten (contracturen). Bij ITB behandeling wordt Baclofen rechtstreeks aan het hersenvocht toegediend. Dit gebeurt door middel van een pompje dat onder de huid van de buik wordt geplaatst, en dat met een onderhuids slangetje is verbonden met het ruggenmerg. Doordat de Baclofen gelijk op de juiste plaats terecht komt, kan het in kleinere hoeveelheden worden gegeven en veroorzaakt het minder bijwerkingen. Op de website www.medtronic.com/nl-nl/
patienten/behandelingen-en-therapieen/baclofenpomp-ernstige-spasticiteit.html kunt u plaatjes van de Baclofenpomp en informatiefolders voor volwassenen en kinderen vinden. Als u vragen heeft over de Baclofenpomp kunt u ook contact opnemen met één van de kinderneurologen van het Amsterdam Leukodystrofie Centrum.
Algemeen
In het Amsterdam Leukodystrofie Centrum wordt ook wetenschappelijk onderzoek gedaan naar leukodystrofieën, juist omdat er nog zo veel vragen zijn. Eén van de ziektes waar wij veel onderzoek naar doen is MLD. Door middel van onderzoek willen wij meer te weten komen over het ziektemechanisme en de klinische en genetische kenmerken van de ziekte. Op dit moment doen wij ook onderzoek verschillende onderwerpen:
- de uitkomst na behandeling(en) bij MLD patiënten
- de MRI afwijkingen bij MLD patiënten
- de zenuwschade bij MLD patiënten
- biomarkers die ons helpen te voorspellen wanneer de ziekte een duidelijke stap achteruit maakt
- de verschillende vormen van MLD en de rol van bepaalde genetische foutjes in het ARSA gen
Hiervoor gebruiken wij (indien er toestemming is van de patiënt en/of de ouders) uitkomsten van de onderzoeken die zijn beschreven in sectie 6 en sectie 7. Onderzoek op een overleden persoon, obductie genoemd, is ook een belangrijke manier om wetenschappelijk onderzoek te doen. Obductie wordt altijd verricht door een patholoog, dit is een arts die gespecialiseerd is in deze vorm van onderzoek. Voor obductie wordt apart toestemming gevraagd.
Als u vragen heeft over meedoen aan wetenschappelijk onderzoek, kunt u altijd contact opnemen met één van de kinderneurologen van het Amsterdam Leukodystrofie Centrum. U kunt de contactgegevens van vinden onder het kopje ‘Meer informatie’. Via hen zijn er ook informatiefolders over meedoen aan wetenschappelijk onderzoek beschikbaar.
Samen met andere artsen uit Europa hebben we een register opgezet voor MLD patiënten, de MLD initiative (the MLD initiative - Disease registry for metachromatic leukodystrophy). Hier kunnen patiënten met MLD zich laten registeren, en er worden gegevens verzameld over het beloop van MLD met en zonder behandelingen.
Heeft u na het lezen van deze informatiefolder nog vragen, dan kunt u bellen met het Amsterdam Leukodystrofie Centrum, telefoon (020) 5667508. U kunt ook de website raadplegen voor informatie over het Centrum van Kinderwittestofziekten:
https://www.vumc.nl/zorg/expertisecentra-en-specialismen/centrum-voor-kinderen-met-wittestofziekten.htm.